Comment les génomes viraux alertent sur l’épidémie de l’hépatite C à Lyon
Le séquençage des génomes viraux et les outils de la phylodynamique peuvent aider la santé publique. Une équipe de recherche a permis de quantifier la propagation du virus de l’hépatite C dans la région lyonnaise. Ces travaux sont publiés dans la revue PloS Pathogens.
Une des difficultés majeures dans le suivi des épidémies est que l’on n’en connaît souvent qu’une fraction. C’est le cas pour les virus de l’hépatite C (VHC) ou de l’immunodéficience humaine (VIH) pour lesquels on parle même « d’épidémie cachée ». En effet, beaucoup de personnes porteuses de ces virus ignorent qu’elles le sont. L’analyse des génomes viraux issus des personnes porteuses dépistées permet en partie de pallier à ce problème de suivi. En effet, les milliers de nucléotides d’une séquence génomique contiennent plus d'informations. Pour deux virus isolés de patients différents, plus ces séquences sont ressemblantes, plus les personnes sont proches dans la chaîne de transmission. Avec le VHC, qui est l’un des virus humains qui accumule le plus vite des mutations dans son génome, l’information est particulièrement riche.
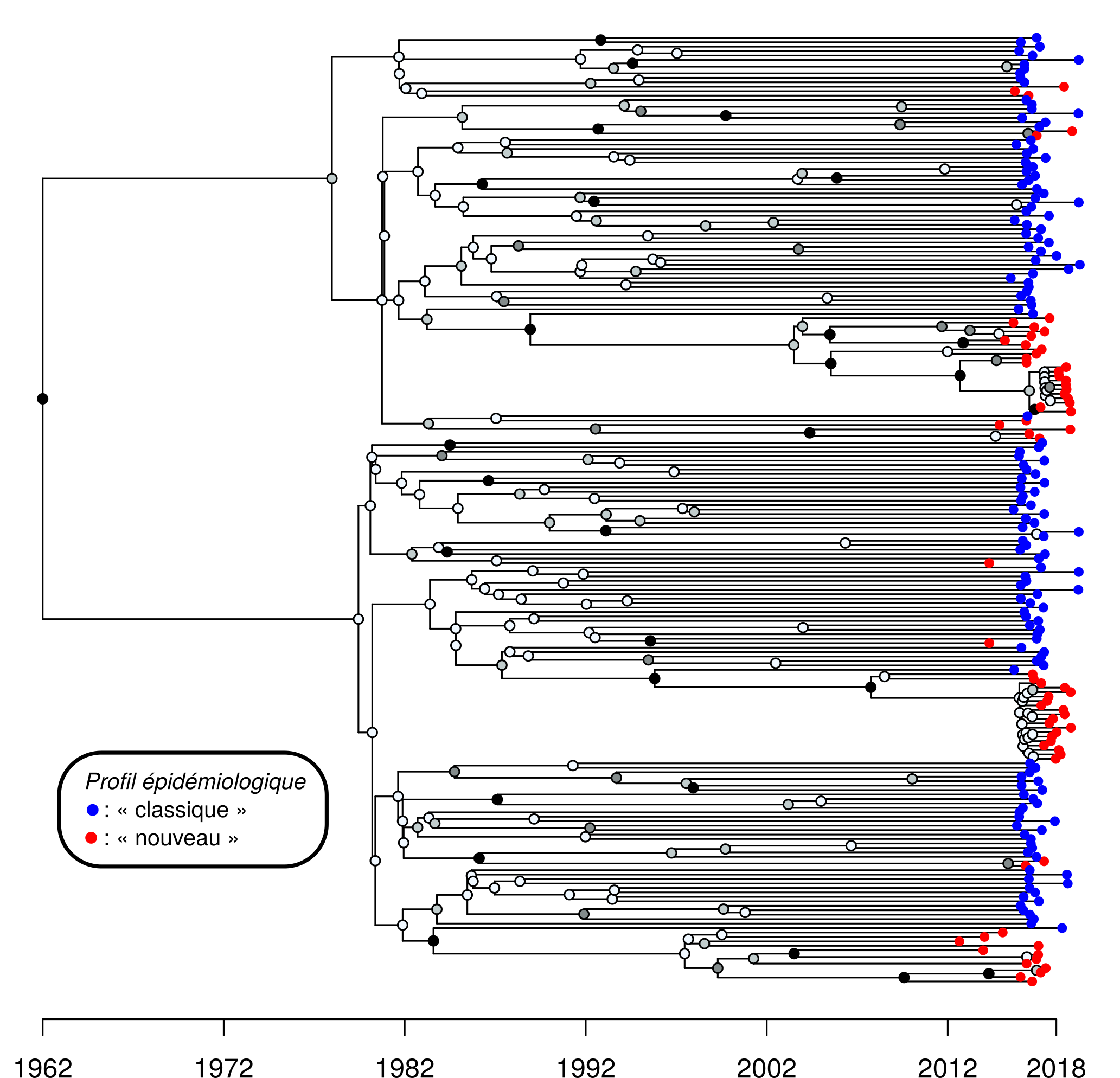
Des chercheurs ont analysé les séquences issues de patients dépistés à Lyon entre 2011 et 2018. Ces séquences reflètent l’existence de deux épidémies : l’une, observée depuis des dizaines d’années, et circulant beaucoup parmi les personnes s’injectant des drogues en intraveineuse et coinfectées par le VIH, l’autre, plus récente, touchant principalement les hommes ayant des rapports sexuels avec des hommes (HSH) et séronégatifs. L’analyse phylodynamique a révélé plusieurs enseignements. D’une part, il existe très peu de contacts entre les deux épidémies. D’autre part, le VHC semble en ce moment se propager bien plus rapidement chez les HSH avec un temps de doublement de l’épidémie de 4 mois environ contre plus de 4 ans pour l’épidémie touchant les consommateurs de drogues. Ces résultats seraient extrêmement compliqués, voire impossibles, à obtenir sans l’information contenue dans les séquences génétiques.
Ces travaux ouvrent plusieurs pistes de recherche pour utiliser au mieux les différents types de données disponibles, c’est-à-dire non seulement les séquences génétiques mais aussi par exemple les données d’incidence. Au niveau de la santé publique, des études complémentaires sur le terrain suivant en priorité des communautés plus à risque permettraient de valider ces résultats. Plus généralement, ces travaux renforcent l’approche consistant à traiter les infections par le VHC dès leur dépistage sans attendre que les patients soient en phase chronique.
Ces travaux ont été soutenus par la fondation pour la recherche médicale (FRM).
Laboratoire du CNRS impliqué
- Maladies infectieuses et vecteurs : écologie, génétique, évolution et contrôle (MIVEGEC - CNRS / IRD / Université de Montpellier)
Objectifs de Développement durable

- Objectif 3 - Bonne santé et bien-être
Référence
Gonché Danesh, Victor Virlogeux, Christophe Ramière, Caroline Charre, Laurent Cotte, Samuel Alizon (2021) Quantifying transmission dynamics of acute hepatitis C virus infections in a heterogeneous population using sequence data. PLoS Pathogens, in press